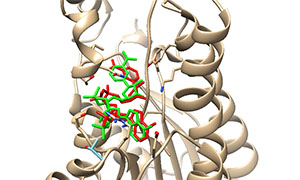
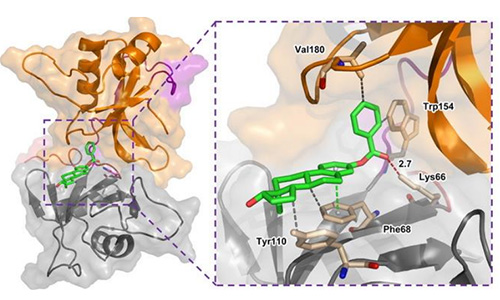
一种基于结构的药物设计方法,通过研究有机小分子配体与生物大分子受体相互作用,预测其结合模式和亲和力。分子对接的本质是两个或多个分子之间的识别过程,涉及分子之间的空间匹配和能量匹配。对接软件将配体小分子放在受体靶标的活性位点处,通过不断优化配体的位置、构象、可旋转键的二面角和受体氨基酸残基的侧链和骨架, 寻找配体小分子与受体靶标结合的最佳构象。
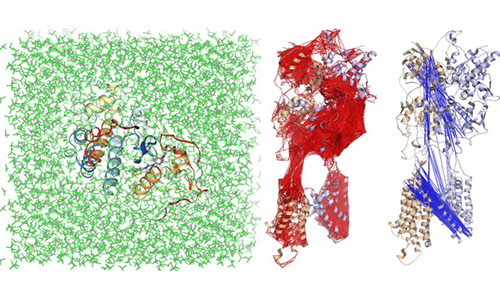
通过分子动力学模拟,研究者得到体系原子的运动轨迹,可观察到原子运动过程的各种微观细节。通过对研究体系的动态模拟,我们能够在分子水平上理解生物大分子的运动与生物功能、蛋白-小分子之间相互作用机理、纳米材料分子的自组装过程。分子动力学模拟是理论计算和实验方法的有力补充,广泛应用于物理、化学、材料科学和生物医药等领域。
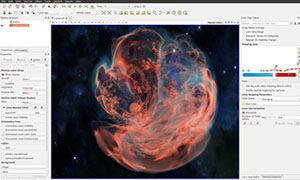
自然界中许多天然化合物分子具有光学手性,正确确定一个有机化合物分子的立体构型,是有机化学工作者尤其是药物研究工作者不可忽视的工作。由于电子圆二色谱(electrostatic circular dichroism, ECD)对分子基团的空间取向非常敏感,能够提供手性分子的三维结构信息,已成为探索手性分子空间绝对构型的有力工具。